北京大学物理学院凝聚态物理与材料物理研究所陈基副教授课题组与字节跳动Seed AI for Science团队、伦敦大学国王学院George H. Booth 教授合作,开发了适用于大规模表面化学计算的多分辨率可系统改进量子嵌入方案,辅以自下而上全流程优化的 GPU 计算框架设计,首次在上万轨道、数百原子的真实表面化学问题的研究中实现“金标准”级的量子化学计算。相关研究成果以 “A multi-resolution systematically improvable quantum embedding scheme for large-scale surface chemistry calculations” 为题,于2025年10月21日发表于《自然-通讯》(Nature Communications) 。
高精度的量子化学计算是化学反应研究、材料和药物设计的重要手段。但是,真正能给出“金标准”级别计算方法通常计算代价庞大,对于包含成千上万电子的表面化学体系难以实际应用。为此,研究团队采用一种能够“分而治之”的多分辨率计算框架(systematically improvable quantum embedding,简称SIE),先用较快、低复杂度的方法在全体系上做初步计算,再将整个系统按照初步计算的结果给出的重要性分割成多个小的区域,最后对这些区域进行更高精度的计算,直到量子化学“金标准”的“CCSD(T)”精度(图1),并把结果再拼接回整体,实现在同一框架内按需平衡速度与精度。“SIE+CCSD(T)”的算法框架通过全面的GPU 优化实现了计算性能的跨越式提升,整体计算复杂度随系统规模近似线性增长,在包含1.1 万个轨道的体系中依然保持高效运行,远超传统“CCSD(T)”计算所能实现的规模。这项工作为未来的材料设计、表面机理探索、乃至高精度高通量计算工作流打开了新的可能性。
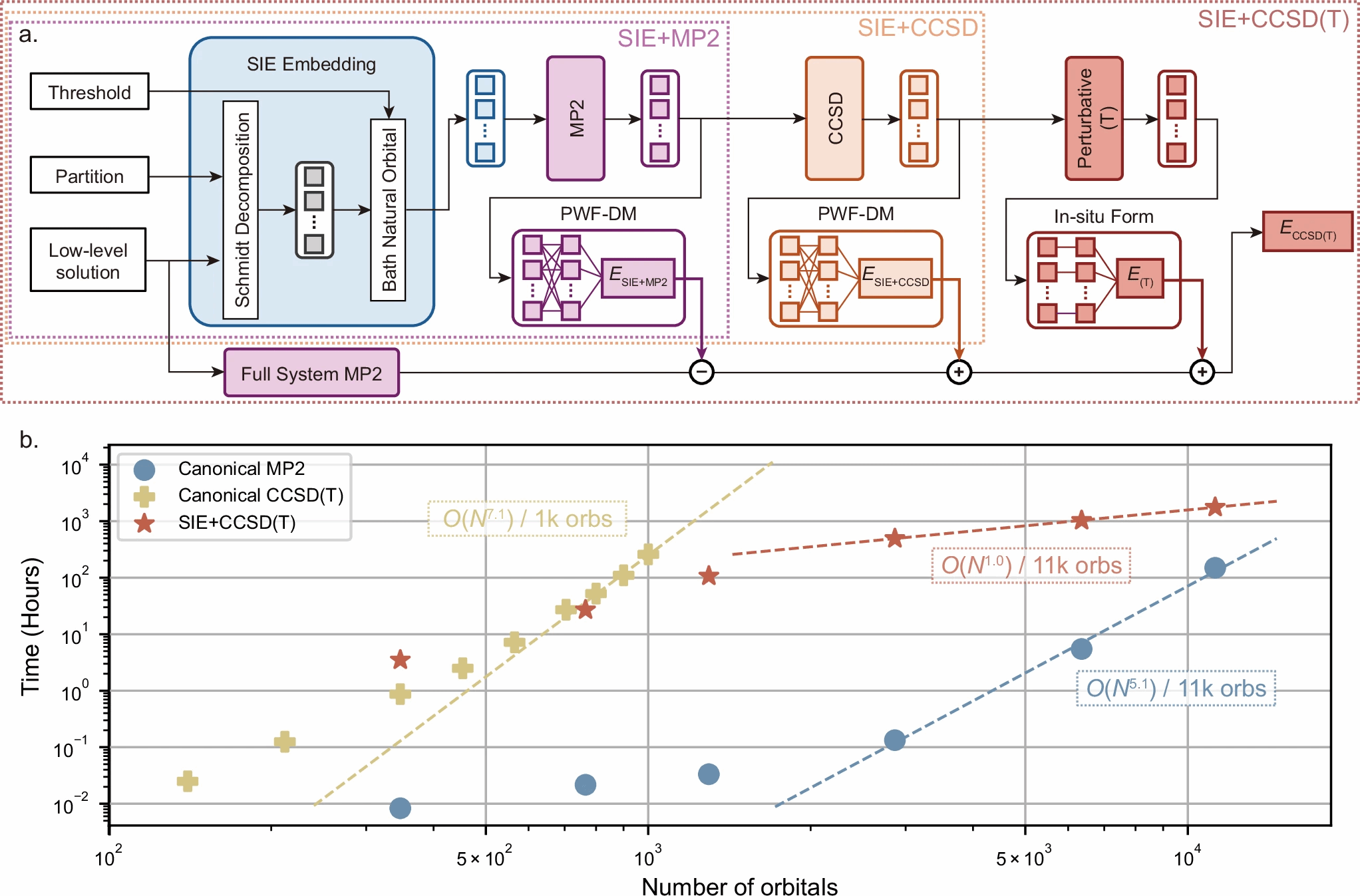
图1: (a)为SIE算法整体框架。(b)展示了各个算法在单A100 GPU卡上计算时随着系统尺寸增长的时间消耗情况。
多结构跨体系的验证
在不同类型的真实体系上,团队用同一套 SIE+CCSD(T) 工作流做了多组测试(图2)。这些体系的结构、化学环境差别很大,但SIE+CCSD(T)计算结果与公开实验数据整体落在 ±1 kcal/mol 的误差范围内。更重要的是,测试中并没有“按体系微调方法”,而是通过统一的策略,在成本可控的情况下把精度稳定地推广到不同材料。这一跨体系的一致性,表明 SIE+CCSD(T) 具备成为“通用工具”的潜力。
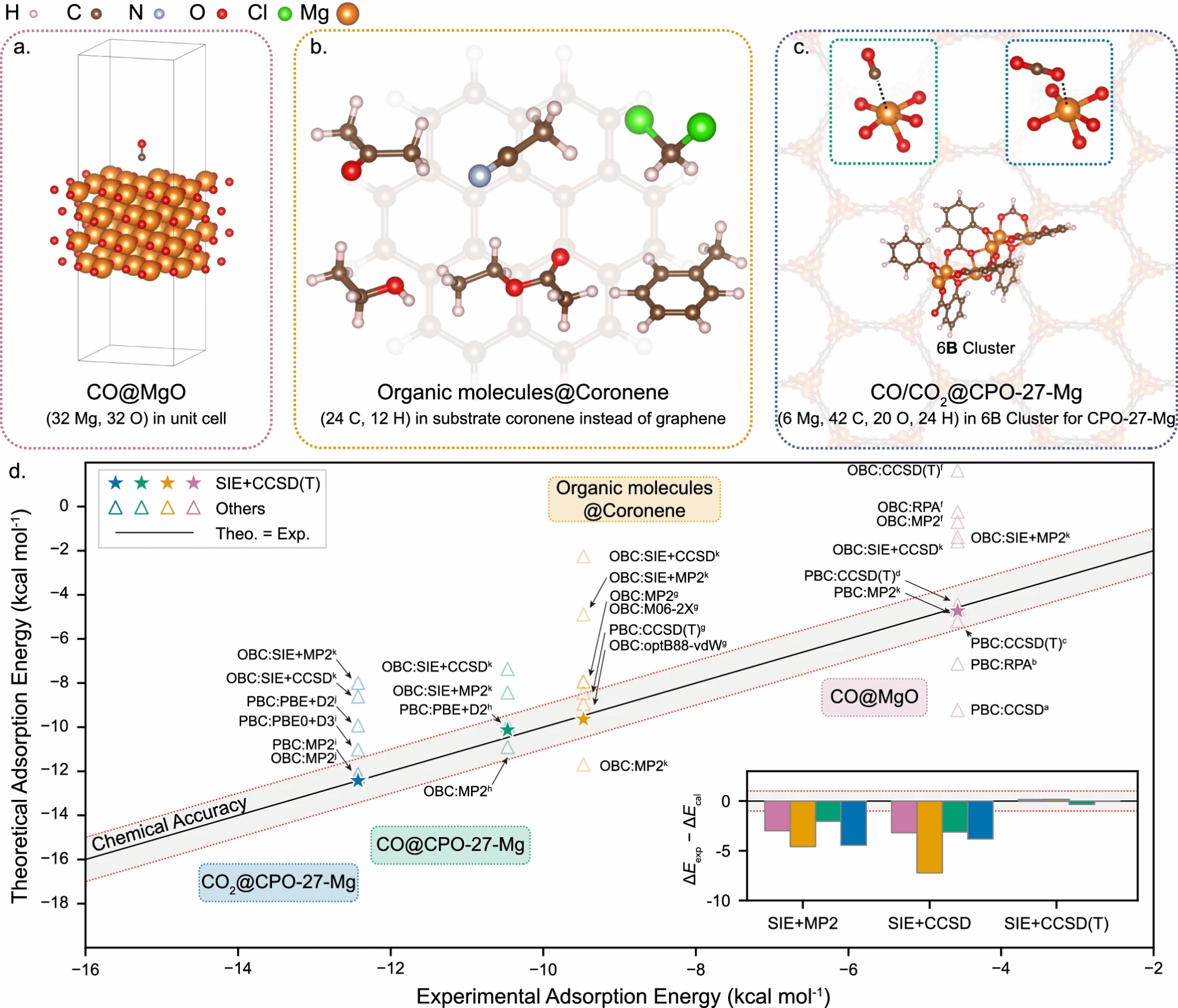
图2:(a)为CO+MgO(001)结构。(b)为CO/CO2+CPO-27-Mg。(c)为有机分子+石墨烯。(d)展示计算结果与实验和其他方法的对比。
澄清石墨烯表面吸附水分子的取向
水分子与石墨烯的相互作用长期以来是表面科学的一个悬而未决的问题,不同研究得出完全相反的结论:有的认为水倾向“趴着”更加稳定,有的认为“竖着”吸附更加稳定。借助 SIE+CCSD(T),研究团队系统地比较了不同水分子取向下的吸附能,发现在小体系下,模拟结果会因边界条件不同而出现巨大差异,而前人的工作中因为缺乏有效计算工具无法再进一步扩大体系尺寸来消除“小体系”的影响。得益于SIE+CCSD(T)的高效性,团队发现当体系扩大到数百原子级别时,开放性与周期性模型的结果逐渐趋同,差距缩小到仅几 meV。最终得到不同取向下的吸附能几乎相同,约为 100 meV。该计算澄清了单个水分子在石墨烯上吸附的基态构型的争论,这将对研究水在石墨烯表面的运动规律提供重要的理论参考。
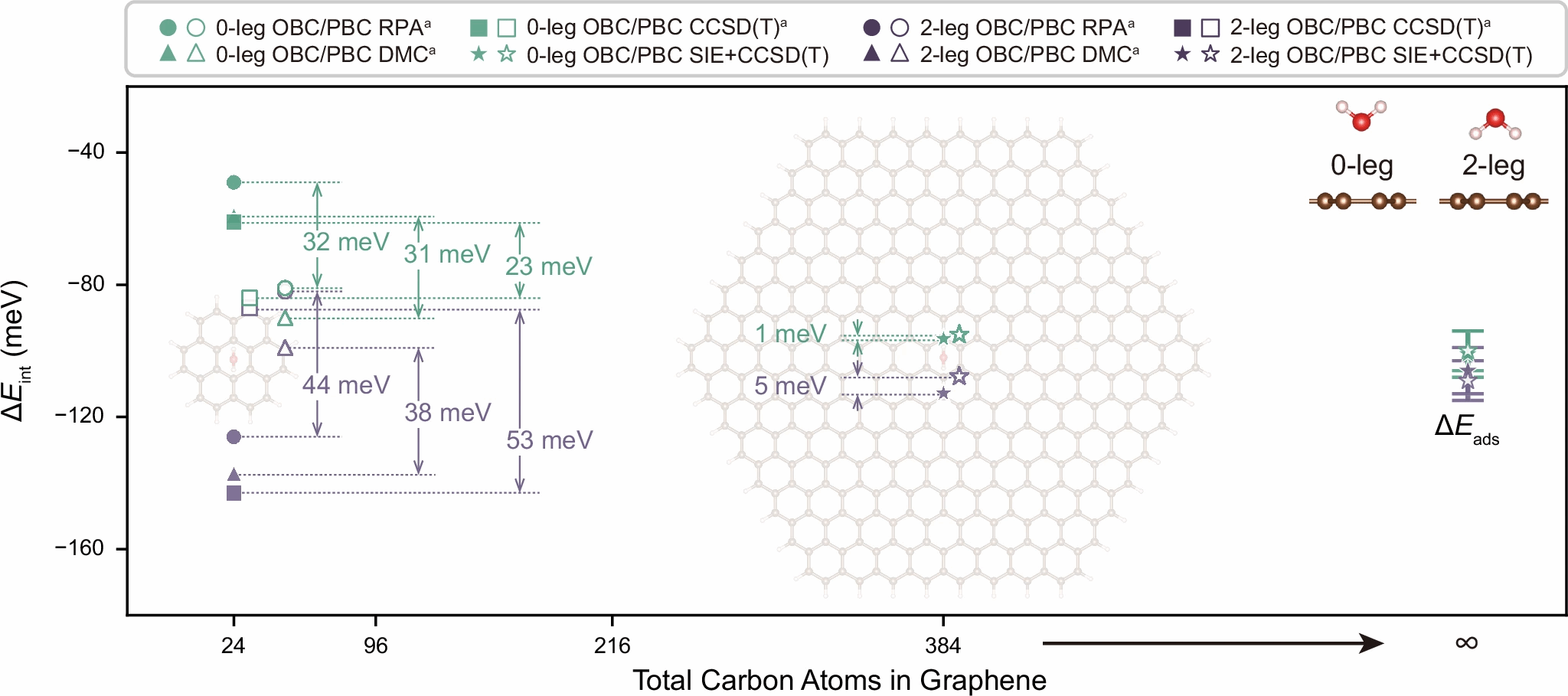
图3:石墨烯表面单个水分子的两种构型的吸附能计算分析。
北京大学物理学院博士毕业生黄子耕(现为字节跳动Seed AI for Science团队研究员)为论文第一作者。黄子耕、George H.Booth、陈基和字节跳动吕定顺研究员为论文共同通讯作者。研究工作得到了国家重点研发计划、国家自然科学基金的资助。
论文原文链接
https://www.nature.com/articles/s41467-025-64374-2


